Lexikon der Chemie: Aldehyde
Aldehyde, zu den Carbonylverbindungen gehörende organische Verbindungen, die eine oder mehrere Aldehydgruppen -CHO im Molekül enthalten. In Abhängigkeit vom organischen Rest, an den die Aldehydgruppe gebunden ist, unterscheidet man aliphatische, aromatische und heterocyclische A.
Nomenklatur. Die Bezeichnung der aliphatischen A. wird nach der IUPAC-Nomenklatur aus dem Namen des entsprechenden Kohlenwasserstoffs, aus dem der Aldehyd formal durch Oxidation ableitbar ist, und dem Suffix -al gebildet:

In der Praxis wird außerdem noch eine ältere Bezeichnungsweise für A. angewendet, die sich vom lateinischen Namen der durch Oxidation des entsprechenden A. gebildeten Carbonsäure ableitet:
;)
Die Bezeichnung aromatischer und heterocyclischer Aldehyde, bei denen die Aldehydgruppe direkt an einem Kohlenstoffatom des betreffenden Ringes gebunden ist, erfolgt vorwiegend durch Trivialnamen, z. B. Benzaldehyd, oder wird aus dem Namen der Stammverbindung und Anfügen von -carbaldehyd gebildet, z. B. Pyrrol-2-carbaldehyd.
Aldehyde. Tab.: Aliphatische und aromatische Aldehyde.
| ||
| Acetaldehyd | CH3-CHO | |
| Propionaldehyd | CH3-CH2-CHO | |
| Butyraldehyd | CH3-(CH2)2-CHO | |
| Acrolein | CH2=CH-CHO | |
| Crotonaldehyd | CH3-CH=CH-CHO | |
| Benzaldehyd | C6H5-CHO | |
| Zimtaldehyd | C6H5-CH=CH-CHO |
Eigenschaften. Die meisten niederen aliphatischen A. sind, abgesehen vom gasförmigen Formaldehyd, leicht bewegliche, stechend riechende Flüssigkeiten. Mit steigender relativer Molekülmasse werden die A. viskoser, die höhermolekularen Vertreter sind fest. Aufgrund des Fehlens intermolekularer Wasserstoffbrückenbindungen liegen die Siedepunkte der A. tiefer als die der entsprechenden Alkohole. In organischen Lösungsmitteln sind A. im allg. gut löslich. Verschiedene A. weisen angenehme, charakteristische Geruchsnoten auf, z. B. Benzaldehyd oder Zimtaldehyd.
|
Reaktionen. Die Reaktionsfähigkeit der A. wird vorwiegend durch die Anwesenheit der Carbonylgruppe/C=O geprägt. Prinzipiell werden bei den für A. typischen Additions- und Kondensationsreaktionen nucleophile Reaktionspartner am partiell positiven Kohlenstoffatom addiert, während elektrophile Reagenzien am partiell negativen Sauerstoffatom gebunden werden. Im Gegensatz zu den meist beständigen Kondensationsprodukten hängt die Stabilität der Additionsprodukte weitgehend von der Natur der Substituenten sowie von den katalytischen Bedingungen ab.
Bei der Addition von Wasser an A. bilden sich Aldehydhydrate (1,1-Diole), die jedoch meist instabil sind, da das Gleichgewicht

gewöhnlich auf der Seite der Reaktanten liegt (Erlenmeyer-Regel). Eine stabile Verbindung dieses Typs ist z. B. Chloralhydrat.
In analoger Weise können in Gegenwart saurer Katalysatoren, wie Halogenwasserstoffe oder Zinkchlorid, Alkohole bzw. Thioalkohole an A. addiert werden. Die dabei gebildeten instabilen Halbacetale reagieren unter den angegebenen Reaktionsbedingungen meist mit einem weiteren Alkohol- bzw. Thioalkoholmolekül unter Wasseraustritt zum Acetal bzw. Thioacetal:
;)
Aufgrund der großen Beständigkeit der Acetale gegenüber Basen werden sie häufig an Stelle der freien A. bei Synthesen eingesetzt. Durch verd. Säuren können Acetale leicht wieder in die Ausgangsverbindungen zerlegt werden.
Durch Reduktionsmittel, wie Wasserstoff, Lithiumaluminiumhydrid und Natriumborhydrid, können A. in primäre Alkohole umgewandelt werden. Durch die Meerwein-Ponndorf-Verley-Reduktion gelingt die Reduktion in Umkehrung der Oppenauer-Oxidation durch Isopropylalkohol in Gegenwart von Aluminiumisopropanolat. Unter den Bedingungen der Wolff-Kishner-Reduktion werden A. reduktiv in die entsprechenden Kohlenwasserstoffe überführt. Die Einwirkung von Oxidationsmitteln auf A. führt zu den entsprechenden Carbonsäuren. Diese Reaktion verläuft außerordentlich leicht, da A. reduzierend wirken. Sie dient deshalb häufig zur Charakterisierung dieser Verbindungsklasse und gleichzeitig zur Unterscheidung von den Ketonen. Als Oxidationsmittel werden dabei Tollens-Reagens, Nylanders Reagens oder Fehlingsche Lösung verwendet.
Eine weitere bekannte Additionsreaktion von A. ist die Anlagerung von Natriumhydrogensulfit unter Bildung schwerlöslicher, kristalliner Hydrogensulfitverbindungen:

Da die Bisulfitaddukte durch verd. Säuren oder Natriumcarbonatlösung leicht wieder in die Ausgangskomponenten zerlegt werden können, dient diese Reaktion häufig zur Reinigung der A.
Eine für zahlreiche Synthesen wichtige Reaktion der A. ist die Bildung von Cyanhydrinen durch Addition von Blausäure an die Carbonylgruppe:
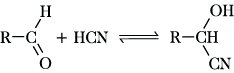
Mit Ammoniak reagieren A. zunächst zu den meist instabilen Aldehydammoniakaddukten, die unter Wassereliminierung leicht in Aldimine übergehen:

Mit primären Aminen kondensieren A. zu Azomethinen:

Bei der Umsetzung mit sekundären Aminen reagieren A. mit α-ständigen Protonen unter Bildung von Enaminen. Bei Abwesenheit des entsprechenden H-Atoms bilden sich Aminale. Durch Grignard-Reaktionen können A. in sekundäre Alkohole umgewandelt werden.
Wichtige Kondensationsreaktionen, die auch bei der Abtrennung und Charakterisierung von A. eine große Bedeutung haben, sind Umsetzungen mit Hydrazin oder substituierten Hydrazinen, z. B. Phenylhydrazin, 4-Nitrophenylhydrazin und 2,4-Dinitrophenylhydrazin, die unter Bildung der entsprechenden Hydrazone ablaufen:

Eine ähnliche wichtige Rolle spielt die Kondensationsreaktion von A. mit Hydroxylamin, bei der Oxime gebildet werden:

Semicarbazone und Thiosemicarbazone entstehen aus A. und Semicarbazid bzw. Thiosemicarbazid. Sie können gleichfalls zur Reinigung und Charakterisierung der A. herangezogen werden.
A. mit α-ständigen Wasserstoffatomen reagieren in Gegenwart von Basen oder Säuren im Sinne der Aldolreaktion.
Aromatische A. disproportionieren unter diesen Bedingungen gemäß der Cannizzaro-Reaktion zu den entsprechenden Carbonsäuren und Alkoholen. In Gegenwart von Aluminiumalkoholaten gelingt diese Disproportionierung auch bei aliphatischen A.
Ähnlich wie bei der Aldolreaktion können A. auch mit anderen CH-aciden Reaktionspartnern, wie Malonsäure und Malonsäurederivaten, gemäß der Knoevenagel-Kondensation, mit Acetanhydrid/ Natriumacetat nach der Perkin-Reaktion sowie mit α-Halogencarbonsäureestern im Sinne der Darzens-Erlenmeyer-Claisen-Kondensation reagieren.
Zahlreiche aromatische A. reagieren unter dem Einfluß von Kaliumcyanid, aber auch einigen Thiazolium- und Imidazoliumsalzen entsprechend der Benzoinkondensation unter Bildung von Benzoinen (Acyloinkondensation). Die strukturanalogen aliphatischen Verbindungen, die Acyloine, können unmittelbar aus aliphatischen A. nur durch Einwirkung von Enzymen bestimmter Hefearten erhalten werden:

Analytisches. Neben der chemischen Charakterisierung in Form von Derivaten können A. und Ketone durch IR- und NMR-spektroskopische Methoden sowie durch massenspektrometrische Fragmentierung charakterisiert werden. Sie weisen in den IR-Spektren intensive Banden für die C=O-Valenzschwingung im Bereich von 1680 bis 1740 cm-1 auf. Die C-H-Valenzschwingungen der A. erschienen bei 2665 bis 2880 cm-1. Die Signale der Aldehydprotonen sind in den 1H-NMR-Spektren im Bereich von δ = 9 bis 10 ppm zu erwarten. In den 13C-NMR-Spektren weisen Signale im Bereich von δ = 180 bis 210 ppm mit hoher Wahrscheinlichkeit auf die C=O-Gruppierung von A. und Ketonen hin. In den Massenspektren von aromatischen A. und Ketonen tritt als charakteristisches Schlüsselbruchstück das Benzoylkation auf.
Vorkommen und Gewinnung. Verschiedene A. kommen in der Natur als Pflanzeninhaltsstoffe in geringen Konzentrationen vor allem in zahlreichen ätherischen Ölen vor, z. B. Citral, Citronellal. Für die Synthese der A. existieren neben zahlreichen, auch für Ketone allgemein anwendbaren Darstellungsmethoden spezielle Verfahren. Am bekanntesten und wichtigsten für die Synthese aliphatischer A. ist die partielle Oxidation oder Dehydrierung von primären Alkoholen:

Als Oxidationsmittel eignen sich Chrom(VI)-oxid oder Kaliumdichromat in Schwefelsäure, Sauerstoff in Gegenwart von erhitztem Kupfer oder Silber, Braunstein sowie Selendioxid. Ein weiteres wichtiges technisches Verfahren ist die Pyrolyse von Gemischen einer Carbonsäure und Ameisensäure in Gegenwart von Mangan(II)-oxid:
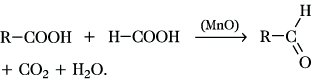
Aliphatische A. können außerdem durch Glycolspaltung sowie durch Hydroformylierung von Alkenen mit Kohlenmonoxid und Wasserstoff in Gegenwart von Dicobaltoctacarbonyl bei etwa 150 °C und etwa 3·104 kPa hergestellt werden. Weitere Aldehydsynthesen sind die Rosenmund-Reduktion von Carbonsäurechloriden, die Grignard-Reaktion von Orthoameisensäureestern mit dem Grignard-Reagens, die Sommelet-Reaktion von Alkylhalogeniden mit Urotropin, die Stephen-Reduktion von Nitrilen, die Nef-Reaktion von primären Nitroalkanen mit verd. Mineralsäuren, die Kröhnke-Reaktion aus Nitronen, die Vilsmeier-Haack-Reaktion von Arenen, die Gattermann-Reaktion von Arenen mit Blausäure und Chlorwasserstoff in Gegenwart von Aluminiumchlorid sowie die Gattermann-Koch-Reaktion.
Verwendung. A. werden hauptsächlich als Ausgangskomponenten für zahlreiche, auch technisch bedeutsame Synthesen eingesetzt sowie als Geruchs- und Geschmacksstoffe in der Lebensmittel- und Kosmetikindustrie. Weit umfangreicher ist das Einsatzgebiet der A. als Zwischenprodukte für die technische Synthese von Kunststoffen, Styryl- und Azomethinfarbstoffen sowie vieler organischer Stoffklassen, z. B. Carbonsäuren, Alkohole, Nitrile, Amine.





Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.