Lexikon der Chemie: Substitution
Substitution, in chemischen Verbindungen der Ersatz eines oder mehrerer Atome bzw. einer oder mehrerer Atomgruppen durch andere Atome oder Atomgruppen. In organisch-chem. Verbindungen ist es oft die S. von H-Atomen, aber auch anderer Atome (Halogene) oder Gruppen (-SO3H), die zu einer Vielzahl von Substitutionsprodukten bei aliphatischen, aromatischen oder heterocyclischen "Stammverbindungen" geführt hat. Die meist gut untersuchten Reaktionsmechanismen sind für typische Stoffklassen von Kohlenstoffverbindungen relevant.
Man unterscheidet radikalische S. (SR), bei denen das die Reaktion einleitende Reagens ein Radikal ist (radikalische Reaktionen), von ionischen Reaktionen wie nucleophile S. (SN), ausgelöst von einem nucleophilen Reagens (nucleophile Reaktionen) bzw. elektrophile S. (SE), ausgelöst von einem elektrophilen Reagens (elektrophile Reaktionen).
Radikalische Substitution. Die SR-Reaktionen laufen bevorzugt bei Alkanen bzw. an Alkylsubstituenten von Arenen oder auch Heterocyclen ab (Alkane). Charakteristisch ist, daß radikalische Bedingungen wie hohe Temperaturen, Licht-, besonders UV-Bestrahlung und Einsatz von Radikalbildnern z. B. die Halogenierung von Toluol in der Seitenkette (-CH3) bewirken, d. h. man erhält Benzylchlorid, Benzalchlorid oder Benzotrichlorid, wobei Kettenreaktionen ablaufen. Durch Wahl der Reaktionsbedingungen können die Reaktionen gelenkt werden. Eine triviale Faustregel ist die S-S-S-Regel, die besagt, daß die oben genannten Reaktionen in der Siedehitze und bei Sonnenlichtbestrahlung in der Seitenkette eintreten, während der Benzolkern nicht angegriffen wird. SR-Reaktionen laufen dann als Kettenreaktionen ab (z. B. radikalische Polymerisation von Alkenen), wenn die Reaktionsenthalpie negativ ist (exotherme Reaktionen). Technisch wichtige SR-Reaktionen sind Halogenierungen, die Sulfochlorierung, die Sulfoxidation, die Nitrierung und die Autoxidation von Alkanen, wobei die Reaktivität bei tertiären C-Atomen besonders hoch ist und zu sekundären und primären C-Atomen hin abnimmt.
Nucleophile Substitution. SN-Reaktionen laufen bevorzugt bei Alkylhalogeniden ab und werden durch Nucleophile wie Br-, I- (Finkelstein-Reaktion), OH- oder OR- (Williamson-Synthese), CN- (Kolbesche Nitrilsynthese), HS- oder RS- (Bildung von Thiolen und Thioethern) oder auch NH3 oder Aminen eingeleitet. Man unterscheidet die monomolekulare SN1-Reaktion von der bimolekularen SN2-Reaktion.
Die SN1-Reaktion folgt einem Geschwindigkeitsgesetz 1. Ordnung. Der geschwindigkeitsbestimmende Schritt ist die Heterolyse einer C-X-Bindung unter Bildung eines Carbenium-Ions, das als Zwischenprodukt nachweisbar ist und mit dem Nucleophil zum Produkt abreagiert:
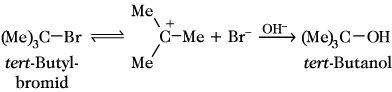
Sterische und elektronische Effekte sowie Stabilisierung des Carbenium-Ions durch Lösungsmittel sind entscheidend für den Ablauf der Reaktion als SN1-Reaktion. Der Angriff des Nucleophils kann von beiden Seiten an dem eben gebauten Carbenium-Ion erfolgen, so daß bei optisch aktiven Edukten eine weitgehende Racemisierung erfolgt.
Die SN2-Reaktion läuft nach einem Synchronmechanismus ab; das Reagens, z. B. CH3O-, nähert sich dem Substrat von der "Rückseite" bezüglich der zu lösenden Bindung C-X. Bindungslösung und Bindungsneuknüpfung erfolgen synchron, wobei ein charakteristischer Übergangszustand durchlaufen wird:
;)
Der geschwindigkeitsbestimmende Schritt gehorcht einem Geschwindigkeitsgesetz 2. Ordnung, mit der Konzentration des Nucleophils steigt die Reaktionsgeschwindigkeit. Die optische Aktivität von Enantiomeren bleibt prinzipiell erhalten, es erfolgt jedoch eine Umkehr der Konfiguration (Walden-Umkehr).
Während die Aktivierungsenthalpien niedriger als bei SN1-Reaktionen liegen, sind die Aktivierungsentropien wegen relativ hoher sterischer Anforderungen an den Übergangszustand von SN2-Reaktionen negativ.
Nucleophile S. hängen von der Konformation des Substrates ab. Die S. von Atomen oder Atomgruppen am Brückenkopf bicyclischer Systeme kann nur nach einem SN1-Mechanismus ablaufen, aber auch dann relativ schwer, weil das Carbenium-Ion wegen der auftretenden Ringspannung keine vollkommen ebene Konfiguration haben kann. Bei Cyclohexylverbindungen werden sowohl in SN1- als auch SN2-Reaktionen axiale Atome oder Atomgruppen wegen des sterisch weniger gehinderten Angriffs des nucleophilen Reagens leicht substituiert. Stereochemische Besonderheiten werden auch bei der Umsetzung primärer und sekundärer Alkohole mit Thionylchlorid beobachtet. Im ersten Reaktionsschritt entsteht ein Alkylchlorsulfit, aus dem sich ein inneres Ionenpaar bildet. Unter Retention der Konfiguration entsteht das Reaktionsprodukt. Der Mechanismus wird als "innere" nucleophile S. (SNi) bezeichnet. Nucleophile Reagenzien mit Ambidenz reagieren entsprechend der Kornblum-Regel.
Je nach dem Abspaltungsmechanismus der austretenden Gruppe unterscheidet man zwischen A1-Mechanismus, vorwiegend bei SN1-Reaktionen und A2-Mechanismus bei SN2-Reaktionen. Im ersten Fall wird eine protonierte Zwischenverbindung (Hydroxy- oder Alkoxyverbindung) monomolekular gespalten:

Beim A2-Mechanismus wird die protonierte Zwischenstufe mit Hilfe des nucleophilen Reagens gespalten (bimolekularer Ablauf).
Nucleophile S. der Allylhalogenide können unter Allylumlagerung verlaufen (SN1'- bzw. SN2'-Substitution).
;)
Speziell bei aromatischen und heterocyclischen Verbindungen verlaufen S. nach folgenden drei Mechanismen:
- SN1-Mechanismus, z. B. beim Verkochen von Arendiazoniumsalzen.
- Additions-Eliminierungs-Mechanismus. Dieser Mechanismus gilt für SN2-S., wobei das angreifende nucleophile Reagens eine Bindung mit dem Reaktionszentrum ausbildet, bevor die Bindung zur Abgangsgruppe gelöst wird. Beispiele hierfür sind die S. der CH3-Gruppe im 2,4,6-Trinitroanisol durch -OR, die Alkalischmelze von Arensulfonsäuren (S. von -SO3H durch -OH) und nucleophile S. am Pyridin. Nach diesem Mechanismus verlaufen auch die meisten Veresterungen und die saure Hydrolyse von Carbonsäureestern sowie die nucleophile S. von Vinylhalogeniden und β-Halogenvinylcarbonylverbindungen.
- Eliminierungs-Additions-Mechanismus. Hiernach verlaufen nucleophile S. an benzoiden Verbindungen (cine-Substitution), Vinylhalogeniden, β-Halogencarbonylverbindungen und β-Halogencarbonsäuren mit stark basischen Nucleophilen, wobei zunächst die zu substituierende Gruppierung und ein Atom oder eine Atomgruppe von einem benachbarten C-Atom unter Ausbildung einer Mehrfachbindung abgespalten wird. An das ungesättigte Intermediat addiert sich eine nucleophile Verbindung.
Elektrophile Substitution. SE-Reaktionen laufen bevorzugt bei Aromaten ab und folgen einem Geschwindigkeitsgesetz 2. Ordnung. Der benzoide Ring wird von einem elektrophilen Reagens, das ein Kation oder ein dipolares Molekül sein kann, unter Ausbildung eines π-Komplexes angegriffen. In einem 2. Reaktionsschritt geht das elektrophile Reagens mit einem bestimmten Ring-C-Atom eine σ-Bindung ein, wobei ein Carbenium-Ion gebildet wird, dessen positive Ladung über das verbleibende konjugierte System delokalisiert ist (σ-Komplex). Durch Abspaltung eines H-Atoms als Proton (normale elektrophile S.) unter Mitwirkung einer Base oder Abspaltung eines anderen Atoms oder einer Atomgruppe (ipso-Substitution) stabilisiert sich der σ-Komplex unter Ausbildung des aromatischen Systems (Substitutionsprodukt). Der SE-Mechanismus ist dem der elektrophilen Addition an Alkene analog, bei letzterer Reaktion stabilisiert sich der σ-Komplex allerdings durch Addition der Base. Als
;)
Base tritt im allg. das bei der Bildung des elektrophilen Reagens entstandene Anion auf.
Das elektrophile Reagens wird in den meisten Fällen in einem der SE-Reaktion vorgelagerten Gleichgewicht gebildet, z. B. bei der Nitrierung mit Salpetersäure HONO2 + HONO2![]()
H2O + NO2+ + NO3- bzw. mit Nitriersäure HNO3 + 2 H2SO4![]()
> NO2+ + H3O+ + 2 HSO4-, bei der Sulfonierung freies SO3 oder HSO3+-Kationen aus H2SO4 + SO3![]()
H2S2O7![]()
H+ H2SO4 + HO-SO2+. Weitere Beispiele für SE-Reaktionen sind Halogenierung, Friedel-Crafts-Alkylierung und -Acylierung, Gattermann- und Gattermann-Koch-Synthese, Houben-Hoesch-Reaktion, Vilsmeier-Haack-Reaktion, Hydroxy- und Chlormethylierung (Blanc-Reaktion), Aminomethylierung, Kolbe-Schmitt-Reaktion, Nitrosierung, Azokupplung, die Mercurierung, die oxidative Kupplung unter Bildung von Indaminen und Indophenolen und die sauer katalysierten Reaktionen von Benzolderivaten mit Aldehyden oder Ketonen (Darstellung von Triphenylmethanfarbstoffen).
Bei der Zweitsubstitution unterscheidet man die vorhandenen Gruppen: Substituenten 1. Ordnung, z. B. CH3- im Toluol, -NH2 im Anilin oder -OH im Phenol erleichtern die Zweitsubstitution und dirigieren den Zweit- oder Drittsubstituenten in o- oder p-Position, wobei vielfach Isomerengemische gebildet werden. Toluol bildet relativ leicht Trinitrotoluol (TNT), Phenol kann in o- oder p-Nitrophenol, 2,4-Dinitrophenol oder 2,4,6-Trinitrophenol (Pikrinsäure) übergeführt werden. Substituenten 2. Ordnung (-NO2, -CHO, -COOH u. a. ungesättigte Gruppen) erschweren die Zweitsubstitution und dirigieren in die m-Position. So entsteht aus Nitrobenzol m-Dinitrobenzol, aus Benzoesäure 3-Nitrobenzoesäure und unter drastischeren Bedingungen 3,5-Dinitrobenzoesäure (vgl. Substituenten).








Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.