Lexikon der Chemie: Molekülorbitaltheorie
Molekülorbitaltheorie, MO-Methode (Abk. von engl. molecular orbital method), ein quantenmechanisches Näherungsverfahren zur Berechnung der Elektronenstruktur und der Energie von Molekülen, das im wesentlichen von Hund, Mulliken, Lennard-Jones und Hückel (1927-29) entwickelt wurde. Dabei betrachtet man analog dem Zentralfeldmodell der Atome ein Elektron, das sich in einem effektiven Potential der Kerne und der übrigen Elektronen bewegt. Die daraus resultierenden Einelektronenzustände des Moleküls bezeichnet man als Molekülorbitale (Abk. MO) φ. Sie erstrecken sich über das gesamte Molekül und sind durch eine Orbitalenergie gekennzeichnet. Die Besetzung der energetisch niedrigsten Molekülorbitale unter Beachtung des Pauli-Prinzips führt zur Elektronenkonfiguration der Moleküle im Grundzustand. Die mathematische Darstellung der Molekülorbitale φ als Linearkombination von Atomorbitalen χi stellt einen geeigneten Näherungsansatz dar (MO-LCAO-Ansatz, Abk. von molecular orbitals – linear combination of atomic orbitals):
.
Beim Variationsverfahren werden die Koeffizienten ci so gewählt, daß die Energie des Gesamtsystems einen Minimalwert annimmt. Das Prinzip der M. läßt sich am einfachsten für homonucleare zweiatomige Moleküle, z. B. für das H2-Molekül, demonstrieren. Die Kombination der 1s-Orbitale der beiden Wasserstoffatome führt zu zwei Molekülorbitalen φ± = N± (χa ± χb), wobei N± den Normierungsfaktor der Plus- bzw. Minuskombination bezeichnet. Die entsprechenden Orbitalenergien ε+ und ε- weisen gegenüber der Energie der Atomorbitale eine niedrigere bzw. höhere Energie auf. Daher bezeichnet man φ+ als bindendes und φ- als lockerndes (antibindendes) Molekülorbital. Im Grundzustand des Wasserstoffmoleküls besetzen beide Elektronen das bindende Molekülorbital (Abb. 1). Damit kann die chemische Bindung im Wasserstoffmolekül erklärt werden. Die Bindungsenergie entspricht dem doppelten Wert von Δε. Das Ausmaß der Aufspaltung der Orbitalenergien ist der Größe der Überlappung der beteiligten Atomorbitale proportional. Mathematisch wird die Überlappung zwischen den Atomorbitalen χa, χb
durch das Überlappungsintegral Sab = ∫ χaχb dv ausgedrückt. Die Überlappung ist groß, wenn die Atomorbitale im gleichen Raumgebiet wesentliche Beiträge aufweisen. Analog zu den 1s-Orbitalen können auch andere Atomorbitale zu Molekülorbitalen kombinieren.
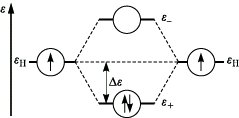
Molekülorbitaltheorie. Abb. 1: Energie der Molekülorbitale für H2.
Aufgrund der durch die Winkelteile (Atommodell) bestimmten Symmetrie und Richtungsabhängigkeit der Atomorbitale unterscheidet man verschiedene Überlappungsmöglichkeiten. Die Kombination von rotationssymmetrisch zur Bindungsachse angeordneten Orbitalen liefert σ-Molekülorbitale. Sie sind durch ein Überlappungsgebiet in der Bindungsachse gekennzeichnet. Die Wechselwirkung von Atomorbitalen, die eine Antisymmetrieebene in der Bindungsachse aufweisen, führt zu π-Molekülorbitalen. Sie bilden zwei Überlappungsgebiete außerhalb der Bindungsachse, die durch eine Knotenebene getrennt sind. Treten vier Überlappungsgebiete außerhalb der Bindungsachse auf, die durch zwei senkrecht zueinander stehende Knotenebenen getrennt sind, so liegt ein δ-Molekülorbital vor. Die so gebildeten Bindungen werden als σ-, π- bzw. δ-Bindung bezeichnet. Die jeweils lockernden Molekülorbitale werden zusätzlich durch einen Stern markiert, z. B. σ*, π*, δ*. Sie weisen neben den Knotenflächen der beteiligten Atomorbitale noch eine zusätzliche Knotenfläche zwischen den Atomen senkrecht zur Kernverbindungsachse auf. Einige Kombinationen von Atomorbitalen zu Molekülorbitalen sind in Abb. 2 angegeben. Bei der Kombination von zwei Atomorbitalen mit unterschiedlicher Symmetrie bezüglich der Bindungsachse ist das Überlappungsintegral Null, und sie können nicht gemeinsam an einem Molekülorbital beteiligt sein (Abb. 3). Ausgehend von den Überlappungsmöglichkeiten der Atomorbitale kann die Molekülorbitalbehandlung auf homonucleare zwei atomige Moleküle von Elementen der 2. Periode (Li2, B2, C2, N2, O2, F2) ausgedehnt werden.
;)
Molekülorbitaltheorie. Abb. 2: Kombinationen von Atomorbitalen zu Molekülorbitalen.

Molekülorbitaltheorie. Abb. 3: Überlappung aus Symmetriegründen Null (S = 0).
Legt man die Kernverbindungsachse als z-Richtung fest und berücksichtigt, daß es für qualitative Betrachtungen genügt, nur die Überlappung zwischen Orbitalen gleicher bzw. ähnlicher Energie zu berücksichtigen, so erhält man das in Abb. 4 veranschaulichte Molekülorbitalschema. Die Molekülorbitale werden in der Reihenfolge zunehmender Energie unter Berücksichtigung der Hundschen Regel und des Pauli-Prinzips mit Elektronen besetzt. Danach beträgt für das Sauerstoffmolekül der Gesamtspin S = 1 (Abb. 4),
;)
Molekülorbitaltheorie. Abb. 4: Qualitatives MO-Schema für ein zweiatomiges Molekül am Beispiel von O2.
was mit dem experimentell beobachteten Paramagnetismus des Sauerstoffs übereinstimmt, der durch die klassische Bindungstheorie nicht erklärt werden konnte. Als qualitatives Maß für die Bindungsstärke zweiatomiger Moleküle dient der Bindungsgrad, der sich als Differenz der Anzahl der Elektronenpaare in den bindenden und lockernden Molekülorbitalen aus dem MO-Schema ergibt. Für Sauerstoff beträgt der Bindungsgrad 2, was etwa der Stärke einer Doppelbindung entspricht. Der MO-LCAO-Ansatz kann auch auf kompliziertere Moleküle angewendet werden, wobei der Rechenaufwand mit steigender Anzahl von Atomorbitalen stark zunimmt. Die Molekülorbitale sind über das gesamte Molekül delokalisiert und spiegeln die Molekülsymmetrie wider. Diese symmetriegerechten Molekülorbitale gestatten die Interpretation von Anregungs- und Ionisierungsvorgängen. Es ist jedoch auch eine gleichwertige Beschreibung der Elektronenstruktur des Grundzustandes mit Hilfe lokalisierter Molekülorbitale (Bindungsorbitale) möglich, die dem klassischen Konzept lokalisierter Bindungen Rechnung trägt und durch zahlreiche experimentelle Befunde (Additivitätsprinzip) gerechtfertigt wird. Zur Konstruktion von Molekülorbitalen können auch Hybridorbitale (Hybridisierung) benutzt werden. Die einfachste Näherungsstufe der M. ist die Hückel-Methode. Durch das Roothaan-Hall-Verfahren ist die M. rechentechnisch so ausgebaut worden, daß auf modernen Rechenanlagen MO-Berechnungen auch an größeren Molekülen durchgeführt werden können.







Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.