Lexikon der Chemie: Arene
Arene, aromatische Kohlenwasserstoffe, benzoide Kohlenwasserstoffe, vom Benzol als Grundkörper abgeleitete cyclisch konjugierte Verbindungen, die sich im allg. durch relativ geringen Energieinhalt und hohe Stabilität sowie besonderes Reaktionsverhalten auszeichnen. Entsprechend ihrer Struktur werden A. eingeteilt in einkernige und mehrkernige A. Zu den einkernigen A. gehören Benzol und seine Homologen (Alkylbenzole, z. B. Toluol, Abb. 1). Die mehrkernigen A. werden weiter unterteilt in Kohlenwasserstoffe, in denen zwei oder mehrere Ringe direkt miteinander verbunden sind (z. B. Biphenyl), in Di- und Polyarylalkane (z. B. Diphenylmethan oder Triphenylmethan) und Kohlenwasserstoffe mit anellierten (kondensierten) Ringen, von denen einer oder mehrere benzoid sind (z. B. Naphthalin), Abb. 2.
;)
Arene. Abb. 1: Einkernige Arene.
;)
Arene. Abb. 2: Mehrkernige Arene.
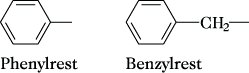
Nomenklatur. Die Bezeichnung der A. erfolgt weitgehend mit Hilfe zugelassener Trivialnamen. Bei mehrfach substituierten Ringsystemen wird die Stellung der Substituenten durch kleinstmögliche Ziffern gekennzeichnet, z. B. 1,2-Dimethylbenzol (o-Xylol). Für die stellungsisomeren Disubstitutionsprodukte sind daneben die Präfixe ortho- (Symbol o-) für 1,2-, meta- (Symbol m-) für 1,3- und para- (Symbol p-) für 1,4- im Gebrauch. Durch formalen Entzug eines H-Atoms aus dem Arenring kommt man zu Arylresten, z. B. Phenyl-, p-Tolyl-, 1-Naphthyl-, die nicht mit dem Benzylrest verwechselt werden dürfen, der durch Abspaltung eines H-Atoms aus der Methylgruppe des Toluols abgeleitet ist (Abb.). Zur Benennung von A. s. auch Nomenklatur, Abschn. C Vl.
Eigenschaften. Einkernige A. sind farblose, leicht entflammbare Flüssigkeiten, die mit stark rußender Flamme brennen. In Wasser sind sie praktisch unlöslich, jedoch gut mischbar mit verschiedenen organischen Lösungsmitteln. Sie sind selbst sehr gute Lösungsmittel, z. B. auch für Fette und Pflanzenöle. Mehrkernige A. sind überwiegend farblose, kristalline Feststoffe, z. T. mit charakteristischem Geruch (Naphthalin). Sie sind wesentlich schwerer entflammbar. Viele von ihnen zeigen im UV-Licht eine charakteristische Fluoreszenz. Aufgrund der besonderen Bindungsverhältnisse und elektronischen Struktur (Aromatizität) weisen die einkernigen A. eine beträchtliche Delokalisierungsenergie auf, die eine hohe Stabilität des aromatischen Ringsystems bedingt. So werden diese Verbindungen im Unterschied zu Alkenen von konz. Schwefelsäure oder Salpetersäure nicht undefinierbar verändert, sondern es kommt zu den für A. typischen elektrophilen Substitutionsreaktionen. Gegenüber Hydrierungsreagenzien sind im allg. nur die benzoiden Ringe inert, d. h., daß z. B. im Naphthalin der eine der beiden Ringe relativ leicht hydriert wird (Tetralin). Im Anthracen und im Phenanthren werden jeweils die mittleren Ringe leicht hydriert bzw. auch oxidiert (Anthrachinon und Phenanthrenchinon).
Reaktionen. Typisch für A. sind elektrophile Substitutionsreaktionen, die unter Erhalt des benzoiden Ringsystems ablaufen (Substitution).
1) Bei der Halogenierung, die in Gegenwart von Eisen(Ill)-halogenid als Katalysator unter schwachem Erwärmen erfolgt, entstehen durch Substitution eines H-Atoms im Arenring Arylhalogenide (Halogenarene). Bei Verwendung eines Halogenüberschusses unter kräftigem Erwärmen können auch zwei oder drei Halogenatome eingeführt werden, die zueinander stets o- oder p-Position einnehmen. Auf diesem Weg können nur Cl- oder Br-Atome eingeführt werden. Bei F- und I-Atomen müssen wegen zu hoher bzw. zu geringer Reaktivität Umwege beschritten werden (Sandmeyer-Reaktion).
2) Die Nitrierung, eine Umsetzung mit konz. Salpetersäure oder einer Mischung von Salpetersäure mit konz. Schwefelsäure (Nitriersäure), erfolgt je nach Reaktivität des A. bei Zimmertemperatur, unter Kühlung oder leichtem Erwärmen. Durch Substitution eines H-Atoms im Arenring entstehen Nitroarene mit einer, zwei oder auch drei Nitrogruppen im Ringsystem.
Toluol reagiert wesentlich leichter als Benzol, daran erkenntlich, daß im Toluol ohne Schwierigkeiten dreifache Nitrierung zu 2,4,6-Trinitrotoluol möglich ist. Im Benzolmolekül sind dagegen nur zwei Nitrogruppen problemlos einzuführen (1,3-Dinitrobenzol).
3) Die Sulfonierung läßt sich mit konz. Schwefelsäure bei Zimmertemperatur oder wenig erhöhter Temperatur durchführen. Durch Substitution eines H-Atoms entstehen stark saure Sulfonsäuren, die auch überwiegend gut in Wasser löslich sind. Allgemein dient die Einführung einer oder auch mehrerer Sulfonsäuregruppen in A. zur Erhöhung der Wasserlöslichkeit vieler Derivate der A., z. B. von Farbstoffen. Außerdem kann die Sulfonsäuregruppe unter Bildung von Sulfonsäurederivaten (Sulfonamide) abgewandelt werden.
4) Bei der Hydroxymethylierung findet eine Umsetzung von A. mit Formaldehyd zu Hydroxymethylarenen C6H5-CH2OH statt. Die Reaktion erfordert bei reaktionsträgen A., wie Benzol, saure Katalysatoren, z. B. Chlorwasserstoff, führt aber dann oft zu Chlormethylarenen, zu mehrfach substituierten Produkten oder auch zu Kondensationsprodukten. Die Umsetzung hat technische Bedeutung zur Herstellung hochmolekularer Kunstharze aus Phenolen und Formaldehyd. Neben Formaldehyd können auch homologe aliphatische Aldehyde und aromatische Aldehyde mit A. im Sinne einer elektrophilen Substitution umgesetzt werden: C6H6 + R-CHO → C6H5-CHOH-R.
5) Bei Formylierungsreaktionen entstehen aromatische Aldehyde, bei denen ein H-Atom des Arenringes durch die Aldehydgruppe ersetzt wird (Gattermann-Synthese, Gattermann-Koch-Synthese und Vilsmeier-Haack-Reaktion). Diese Reaktionen sind jedoch auf besonders reaktionsfähige A. ("aktivierte A.") begrenzt und z. B. am Benzol nicht durchführbar.
6) Die Nitrosierung, eine Umsetzung mit salpetriger Säure, ist ebenfalls nur mit aktivierten A., wie Phenolen oder tertiären aromatischen Aminen, möglich und dient zur Einführung einer Nitrosogruppe in 1,4-Stellung zum aktivierenden Substituenten:
R-C6H5 + HNO2![]()
R-C6H4-NO.
Analog dazu verlaufen auch die Azokupplung mit Diazoniumsalzcn, die Azofarbstoffe ergibt, sowie die Carboxylierung mit Kohlendioxid (Kolbe-Schmitt-Synthese), die zu Carbonsäuren führt, nur mit aktivierten A. in praktisch verwertbaren Ausbeuten.
7) Umsetzung mit Alkylhalogeniden (Friedel-Crafts-Reaktionen).
8) Chlormethylierung (Blanc-Reaktion).
9) Die Substitution eines H-Atoms im Arenring durch nucleophile Reagenzien ist sehr schwierig und spielt praktisch keine Rolle. Dagegen sind SN-Reaktionen an verschiedenen substituierten A. möglich, wenn geeignete Abgangsgruppen vorhanden sind. Dazu gehören Halogenatome, Sulfonsäuregruppen und die Diazoniumgruppe, die leicht Stickstoff abspartet (Sandmeyer-Reaktion).
10) Oxidationsreaktionen sind bei Alkylarenen und anellierten Ringsystemen vielfach möglich, z. B. die Oxidation von Toluol zu Benzoesäure oder p-Xylol zu Terephthalsäure. Dazu werden die bekannten klassischen Oxidationsmittel Chrom(VI)-Verbindungen in Eisessig oder Schwefelsäure bzw. Kaliumpermanganat in alkalischer Lösung verwendet. Technisch werden aromatische Carbonsäuren in großem Umfang durch Luftoxidation in Gegenwart von Vanadium(V)-oxid oder Cobaltsalzen aus Methylbenzolen hergestellt.
A. lassen sich grundsätzlich mit katalytisch aktiviertem Wasserstoff unter Druck und bei erhöhter Temperatur zu gesättigten cyclischen Ringsystemen hydrieren, z. B. Benzol zu Cyclohexan oder Naphthalin zu Decalin (Decahydronaphthalin). Davon wird in der Technik und in wissenschaftlichen Labors zur Gewinnung sonst schwer zugänglicher Verbindungen Gebrauch gemacht. Dabei muß im Autoklaven gearbeitet werden.
Analytisches. A. werden durch Nitrierung und Reaktion mit Aluminiumchlorid und Chloroform (Friedel-Crafts-Reaktionen) nachgewiesen. Die Identifizierung erfolgt entweder durch Sulfochlorierung (Umsetzung mit Chlorsulfonsäure) mit anschließender Aminolyse oder durch Adduktbildung mit Pikrinsäure. A. mit Alkylseitenketten können mit Kaliumpermanganat in alkalischer Lösung zu Carbonsäuren oxidiert werden, einige A., wie Anthracen oder Phenanthren, werden mit Chrom(VI)-Verbindungen zu Chinonen oxidiert. In den IR-Spektren des Benzols und seiner Derivate erscheinen C-H-Valenzschwingungen bei etwa 3030 cm-1 sowie benzoide C=C-Valenzschwingungen bei etwa 1500 und 1600 cm-1. Für die Identifizierung ein- oder mehrfach substituierter Verbindungen ist das Gebiet von 650 bis 850 cm-1 ("out-of-plane" Schwingungen) wichtig. Die UV-Spektren des Benzols und seiner Alkylderivate zeigen drei Absorptionsbanden bei 180 nm, 200 nm und 255 nm (π → π*-Übergang). Bei zahlreichen anellierten Ringen (besonders lineare Anellierung) rückt das langwelligste Absorptionsmaximum in den sichtbaren Spektralbereich. Bathochrome Verschiebung tritt auch bei Einführung bestimmter Substituenten ein, z. B. Nitrogruppen, Nitrosogruppen. Im 1H-Kernresonanzspektrum erscheinen infolge der durch den Ringstrom verminderten Abschirmung die Protonensignale bei δ-Werten von 6,5 bis 8,5 ppm. Aus den Kopplungskonstanten können wichtige Rückschlüsse auf den Substitutionstyp gezogen werden (Jortho = 6 bis 10 Hz, Jmeta= 1 bis 3 Hz, Jpara= 0 bis 1 Hz). In den Massenspektren des Benzols und seiner Derivate tritt jeweils ein starker Molpeak auf. Der Basispeak der Alkylbenzole wird vom Tropylium-Ion C7H7+ (MZ = 91) gebildet. Weitere typische Fragmente erscheinen bei MZ = 77, 65, 53, 51, 50 und 39.
Vorkommen und Gewinnung. Benzol und viele seiner Homologen kommen in Erdölen verschiedener Herkunft, im Kokereigas und im Steinkohlenteer vor, aus dem sie im technischen Maßstab abgetrennt werden. Zur Deckung des steigenden Bedarfs an diesen Kohlenwasserstoffen führt man aliphatische und gesättigte cyclische Bestandteile des Erdöls durch Dehydrocyclisierung, Dehydroisomerisierung, Dehydrierung oder Hochtemperaturcracken in A. über. Viele mehrkernige A. finden sich in beträchtlichen Mengen im Steinkohlenteer oder auch im Teer der Benzin- und Kerosinpyrolyse und werden daraus gewonnen. Daneben gibt es vielfältige Synthesen, die z. T. technisch oder im Labor durchgeführt werden:
1) Alkylbenzole werden durch die Wurtz-Fittig-Reaktion aus Halogenbenzol und Halogenalkan mit Natrium, durch die Friedel-Crafts-Alkylierung aus A., Halogenalkanen und Aluminiumchlorid sowie durch die Wurtz-Grignard-Reaktion aus Arylmagnesiumchlorid und Halogenalkanen erhalten. Bei der vielseitig variierbaren Friedel-Crafts-Alkylierung werden oft anstelle der Halogenalkane die technisch verfügbaren Alkene Ethen oder Propen als Alkylierungsmittel verwendet.
2) Diphenylmethan und Triphenylmcthan sind ebenfalls durch Friedel-Crafts-Reaktionen zugänglich, z. B. durch Umsetzung von Benzol mit Benzylhalogenid bzw. von Benzol mit Chloroform.
;)
Arene. Abb. 3: ortho-Spaltung des aromatischen Ringes und Reaktionen des 3-Ketoadipat-Weges beim mikrobiellen Abbau monocyclischer Aromaten (Enzyme: (1) Catechol-1,2-dioxygenase, (2) Muconatcycloisomerase, (3) Muconolacton-Δ-isomerase, (4) Protocatechuat-3,4-dioxygenase, (5) Carboxymuconatcycloisomerase, (6) Carboxymuconolactondecarboxylase, (7) 3-Ketoadipat-enol-lactonase, (8) 3-Ketoadipat-Succinyl-CoA-Transferase, (9) 3-Ketoadipyl-CoA-Thiolase).
3) Anellierte A. können für präparative Zwecke durch Friedel-Crafts-Acylierung erhalten werden, z. B. Naphthalin aus Benzol und Bernsteinsäureanhydrid oder Anthracen aus Benzol und Phthalsäureanhydrid.
Verwendung. Benzol und seine Homologe, besonders Toluol und die Xylole, werden als organische Lösungsmittel oder Extraktionsmittel verwendet, wobei ihre hohe Giftigkeit (Benzol) beachtet werden muß. Sehr viele aromatische funktionelle Verbindungen haben als Synthesezwischenprodukte große technische und wissenschaftliche Bedeutung.
Mikrobieller Abbau. Neben Bakterien (vor allem Pseudomonaden, Actinomyceten) können auch Hefen bzw. Pilze (Candida, Aspergillus, Penicillium) A. abbauen. Dies stellt einen wichtigen Schritt im Kohlenstoffkreislauf der Natur dar.
Trotz der Vielfalt aromatischer Verbindungen sind folgende einheitliche Abbauprinzipien durch Mikroorganismen erkennbar: 1) Vorbereitung der Ringspaltung durch Einführen von Hydroxygruppen durch Monooxygenasen und Dioxygenasen. Es folgt im allgemeinen eine zweite Hydroxylierung – meist in ortho-Stellung. Die Mehrzahl der Kohlenwasserstoffe wird dabei zu zwei Hauptintermediaten umgewandelt: Brenzcatechin und Protocatechusäure. 2) Ringspaltung: Die aromatischen Diole werden – in Abhängigkeit vom Organismus – durch Dioxygenasen an drei verschiedenen Positionen geöffnet: ortho-Spaltung (Abb. 3), meta-Spaltung und Gentisinsäurespaltung. 3) Abbau der Spaltprodukte zu Intermediaten zentraler Stoffwechselwege. Nach der ortho-Spaltung werden Brenzcatechin und Protocatechusäure über den 3-Ketoadipinsäure-Weg zu Succinat und Acetyl-CoA abgebaut (Abb. 3). Auch bei der meta-Spaltung von Brenzcatechin und Protocatechusäure entsteht nach Abspaltung von Ameisensäure eine Ketosäure, die zu Pyruvat und Acetaldehyd (aus Brenzcatechin) bzw. 2 Mol Pyruvat (aus Protcatechusäure) abgebaut wird.
Der Abbau der kondensierten aromatischen Kohlenwasserstoffe erfolgt Ring für Ring. Wichtigstes Intermediat ist die Salicylsäure, welche teilweise von verschiedenen Bakterien während der Assimilation ins Kulturmedium ausgeschieden wird.
Einfach substituierte aromatische Verbindungen (u. a. Phenol, Mandelsäure, Benzoesäure) und ortho-disubstituierte Verbindungen (z. B. Salicylsäure, Anthranilsäure) werden ebenso wie Naphthalin, Anthracen und Phenanthren über Brenzcatechin abgebaut. o-Cresol, m-Xylen und m-Cresol können zu 3-Methylbrenzcatechin, p-Xylol und p-Cresol zu 4-Methylbrenzcatechin abgebaut und nach meta-Spaltung zu Essigsäure, Acetaldehyd und Pyruvat (3-Methylbrenzcatechin) bzw. Kohlendioxid, Propionaldehyd und Pyruvat (4-Methylbrenzcatechin) weitermetabolisiert werden.
Zum Totalabbau halogenierter Aromaten sind nur wenige Mikroorganismen befähigt. Allgemein verläuft der Abbau halogenierter Derivate aufgrund sterischer und elektronischer Effekte wesentlich langsamer als der der unsubstituierten Analoga. Für die Abbaubarkeit ist sowohl die Anzahl als auch die Position der Halogenatome entscheidend. Die Intermediate sind toxisch, solange das Halogenatom am Molekül gebunden ist. Das Spektrum der mikrobiell abbaubaren halogenierten Aromaten kann durch Cometabolismus und genetisch veränderte Mikroorganismen beträchtlich erweitert werden.







Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.